
SAM is a method that can be used to identify genes that are significantly differentially expressed. Each gene is assigned a score on the basis of its change in gene expression relative to the standard deviation of repeated measurements. The genes that have a score higher than some adjustable threshold are used to estimate the significance of the result. This is done by permuting the measurements to see how many genes comes up with a score above the threshold. The percentage of genes falsely identified as differentially expressed is called False Discovery Rate (FDR).
For more details on the SAM method, see PNAS (Tusher et al. 2001 98: 5116-5121)
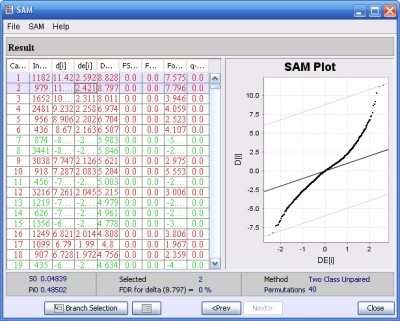
Example of the SAM result window with the two highest scoring genes are selected
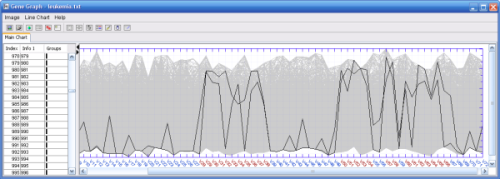
The result above displayed in the line chart window
See how the expression differs in the blue and the red class
