Robust Multi-array Average (RMA)
RMA is an algorithm used to create an expression matrix from Affymetrix data. The raw intensity values are background corrected, log2 transformed and then quantile normalized. Next a linear model is fit to the normalized data to obtain an expression measure for each probe set on each array. RMA is very easy to use in J-Express, a description follows further down on this page. All transformations and normalizations change the data, and it is highly recommended to understand how. For more on RMA, see:
- Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D., Antonellis, K. J., Scherf, U.,
Speed, T. P. (2003). Exploration, Normalization, and Summaries of High Density
Oligonucleotide Array Probe Level Data. Accepted for publication in Biostatistics.
RMAExpress
RMAExpress is a stand-alone program by Bolstad et al, which compiles affymetrix chip data into gene expression data. If you experience problems using RMA in J-Express, then this is a very good alternative to get the gene expression values. You can then import the gene expression data into J-Express using Load Tabular Data from the J-Express File menu
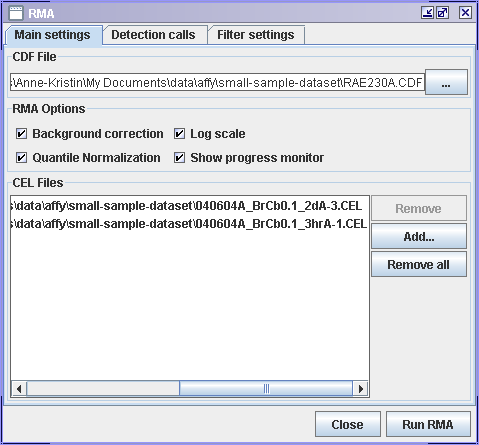
RMA in J-Express
Memory usage:
RMA uses a lot of memory since it works on all arrays at the same time. If you encounter memory problems, you can increase the java heap size. See here how you can edit the J-Express settings.
How to load affymetrix data using RMA:
- Open RMA from File | Load Affymetrix data using RMA
- In the Main settings tab:
- Locate the CDF File. J-Express will automatically look for CEL files in the same folder as the .CDF file and add them to the CEL Files list.
- If .CEL files have been added to the CEL Files list, view the list to see that it contains the ones you want to use. If you need to make any changes to it, use the Remove and Add buttons to get the files you want.
- Select the RMA Options you want to use. Large datasets, e.i. more than 10 .CEL files may take long to process. It is then a good idea to check the Show progress monitor so that you can see that it is working and to monitor memory usage.
- I the Detection calls tab:
- The detection algorithm calculates a score for each probeset that is used to call a transcript present, marginal or absent. The sensitivity and spesificity of the detection algorithm can be adjusted by changing the Alpha and Tau parameters.
- Tau influences the detection p-value, which is used to call a probepair present or absent. Increasing the threshold Tau can reduce the number of false Present calls, but may also reduce
the number of true Present calls.
- Probe sets with detection p-value lower than Alpha 1 are called Present.
- Probe sets with detection p-value higher than Alpha 2 are called Absent.
- Probe sets with detection p-value in-between Alpha 1 and Alpha 2 are called Marginal.
- In the Filter settings tab:
- Minimum percentage of 'Present' genes: means that genes that have less than e.g. 50 % Present calls for all arrays will be removed.
- Maximum percentage of 'Absent' genes: means that genes that have more than e.g. 50 % Absent calls for all arrays will be removed.
- Press Run RMA.
- When RMA has finished, the data will be available in the J-Express Project window.